Research
My research interests cover various topics of microbial
genomics from development of new methodology and their application for medical purposes to fundamental questions of genome organization and evolution.
Phase variation in gut Bacteroides
*A collaboration with David Berry, UniWien and Calin Guet, ISTA. Supported by the FWF grant #ESP 253-B

The goal of the project is to reveal novel phase variable systems in gut microbes. Gut microorganisms thrive in a dynamic environment characterized by simultaneous changes in nutrient availability, phage infection pressure, and host immune system regulation. To cope with this variable environment, gut microorganisms rely on phase variation, which is the rapid emergence of intra-population genomic and phenotypic heterogeneity caused by the reversible switching of gene expression in individual cells. Studying this phenomenon shaped our understanding of gut microbiota ecological dynamics, evolutionary theory, and human health, yet its molecular basis is thought to be species-specific and remains poorly understood. We discovered an evolutionary conserved phase variable system common to Bacteroidaceae, an abundant family in the mammalian gut microbiota. The invertase homologs are inherited in genetic linkage with the machinery that activates diverse silent susCD transporter genes.An analogy of this phase variation system is a jukebox - an automatic record-player in which a mechanical arm selects a single record from a pre-loaded library and places it into a location where it can be played.
Detecting the impact of large-scale genomic features on bacterial phenotypes
*Supported by the FWF grant #ESP 253-B
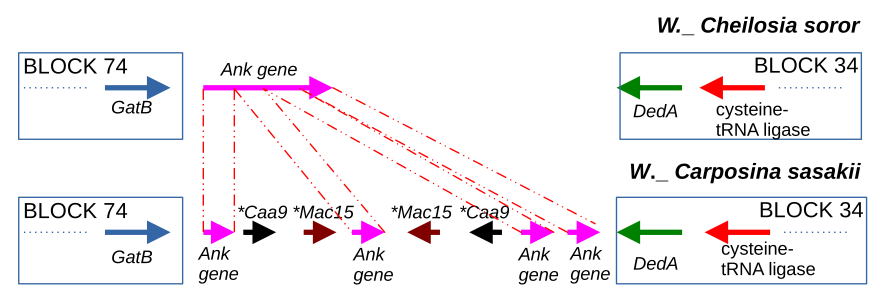
The main goal of the project is to connect the structural variants with phenotypic changes. To date, the important role of genome rearrangements in generating phenotypic variation in bacteria were shown, provoking the high interest of microbiologists to the topic and highlighting the necessity of comparative genomics pipelines to study structural variants in genotype-phenotype context. Representation of microbial genomes as an ordered set of locally collinear blocks (LCBs) is an efficient strategy for revealing structural variants in bacterial genomes on genus, species or colony level of evolution. But not being integrated into comparative genomics pipelines, such tools are not commonly used, as the biological interpretation of their results remains challenging. Here we close this gap by introducing the term ‘pan-blockome’, the whole LCB repertoire of a studied group of genomes, and present the package for pan-blockome genomic studies called BADLON.
Role of gene paralogs in bacterial chromosome maintainance
*Supported by the FWF grant #ESP 253-B
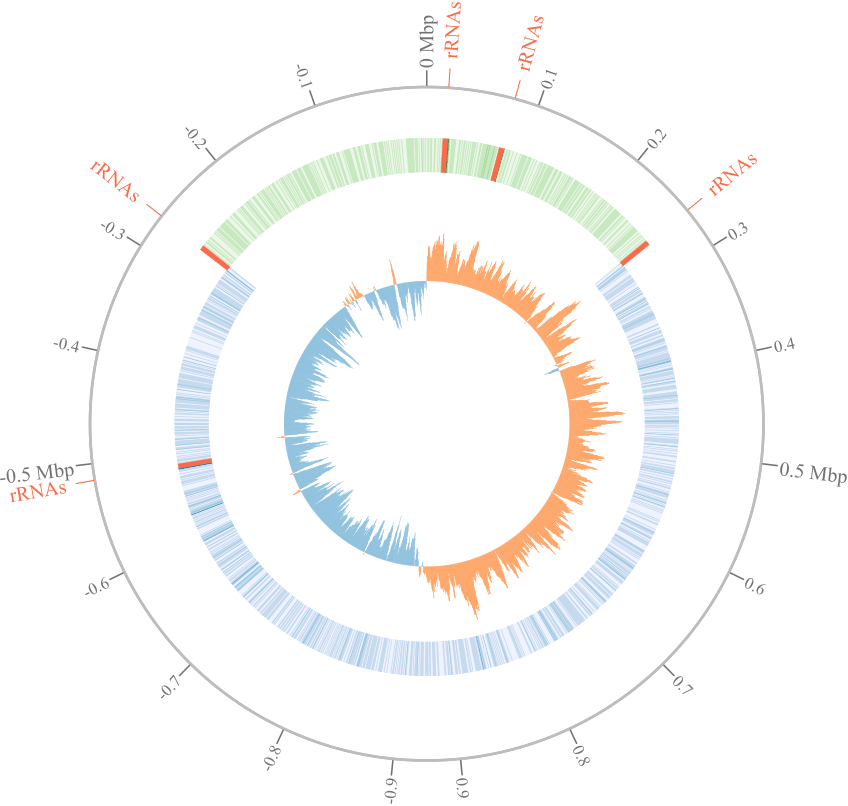
The project aims to investigate the patterns on genomic repeats across the circular bacterial chromosome to reveal interplay between chromosome topology and gene paralogization. Copy number variation is the important genomic trait associated with bacterial phenotype. In particular, number of rRNA gene operons is species-specific and supposed to be associated with ecological niches. On the other hand, such genomic repeats provide substrates for intra-genomic recombination leading to genome rearrangements. We assume that recombination events as well as composition of genomic repeats are shaped by selection forces balancing profit and damages on different levels of chromosome organization.
Role of noncanonical start codons in bacteria
*A collaboration with Calin Guet, IST Austria
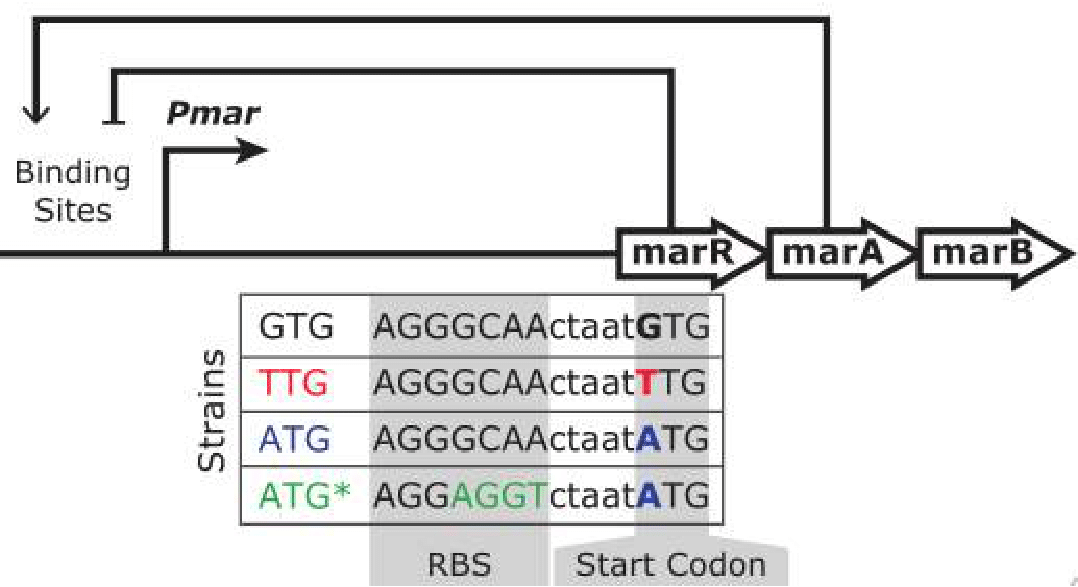
The project is aimed to understand the role of noncanonical, yet highly conserved start codons in bacterial genes Active regulation of gene expression, orchestrated by complex interactions of activators and repressors at promoters, controls the fate of organisms. In contrast, basal expression at uninduced promoters is considered to be a dynamically inert mode of nonfunctional “promoter leakiness,” merely a byproduct of transcriptional regulation. Here, we investigate the basal expression mode of the mar operon, the main regulator of intrinsic multiple antibiotic resistance in Escherichia coli, and link its dynamic properties to the noncanonical, yet highly conserved start codon of marR across Enterobacteriaceae. Real-time, single-cell measurements across tens of generations reveal that basal expression consists of rare stochastic gene expression pulses, which maximize variability in wildtype and, surprisingly, transiently accelerate cellular elongation rates. We uncover a surprising role for this basal expression mode in general growth homeostasis of Enterobacteria that is intimately tied to their ecophysiology. We reveal how selective forces can shape gene expression modes of a global transcriptional regulator by differently trading off evolutionary costs and benefits. Understanding these trade-offs specifically for the mar operon as the main determinant of intrinsic multidrug resistance in Enterobacteria is essential for public health.
Machine learning and phylogenetic analysis for prediction of antibiotic resistance
*A collaboration with Olga Kalinina, HIPS Germany
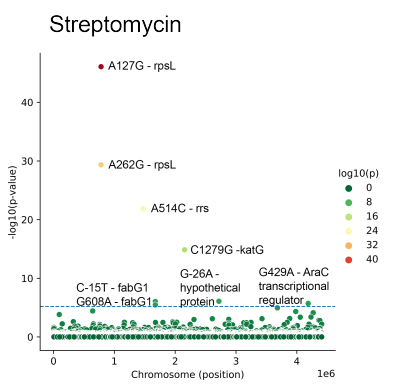
The project is aimed to develop ML models for discovery of antibiotics resistance markers. We present ML models for the discovery of antibiotic resistance markers. The models are trained using whole-genome sequences with accompanying resistance screens, and the resistance markers are extracted with feature importance analysis. We emphasize the importance of accounting for population structure within a bacterial species by introducing PRPS, phylogeny-related parallelism score. We show that ML models that employ PRPS-aware features demonstrate superior performance, as well as discover more biologically meaningful markers.
Fitness effects of short random peptides
*A collaboration with Dan I. Andersson, Uppsala University and Roderich Roemhild, IST Austria

The project is aimed to estimate the distribution of fitness effects of random non-coding DNA in microbial cells. It is generally assumed that new genes arise through duplication and/or recombination of existing genes. Previous experimental work confirmed that new functional genes could arise out of random non-coding DNA. In our research we estimate the fitness effects of de-novo transcribed random DNA in E. coli.
Development of bioinformatic tools for detection and analysis of large-scale genomic variants in bacterial genomes
*A collaboration with Nikita Alexeev, ITMO
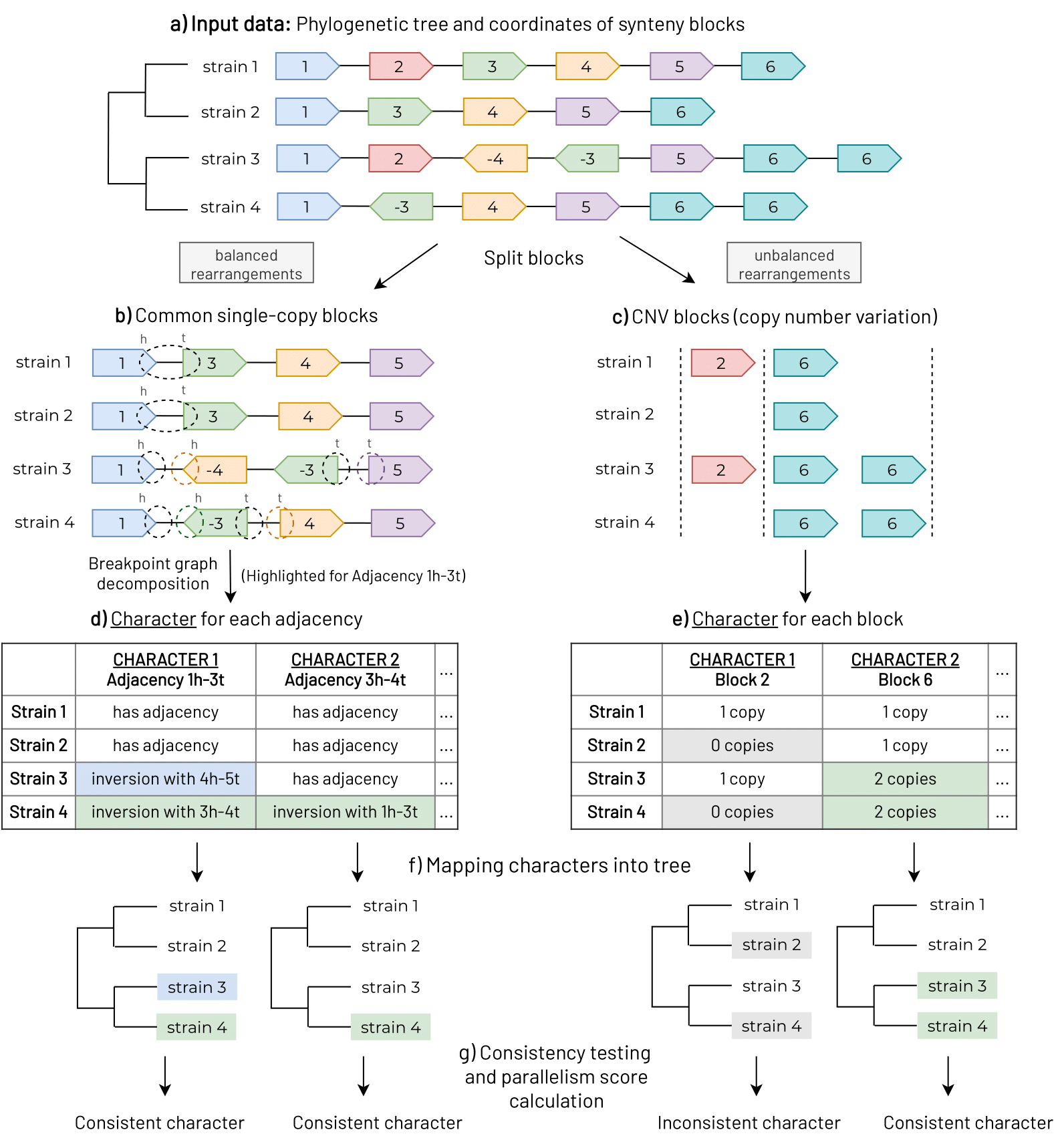
The project aims to develop bioinformatic software to detect large-sclae genomic variants in bacterial genomes and linked them with bacterial phenotypes. We have developed a strategy for extract information form genomic data to detect parallel rearrangements in bacterial populations. The approach will be used for the study of rapid emergence of new bacterial phenotypes, understanding the molecular basis of antibiotic resistance mechanisms and formation of small colony variants, and the study of the selective forces in genomic evolution underlying complex phenotypes. The application of this approach and the concomitant understanding of connections between detected genome rearrangements and medically-relevant phenotypes may contribute to the efficient development of drugs and vaccines.
Origin and evolution of the multi-chromosome bacterial genomes
*A collaboration with Mikhail Gelfand, Skoltech
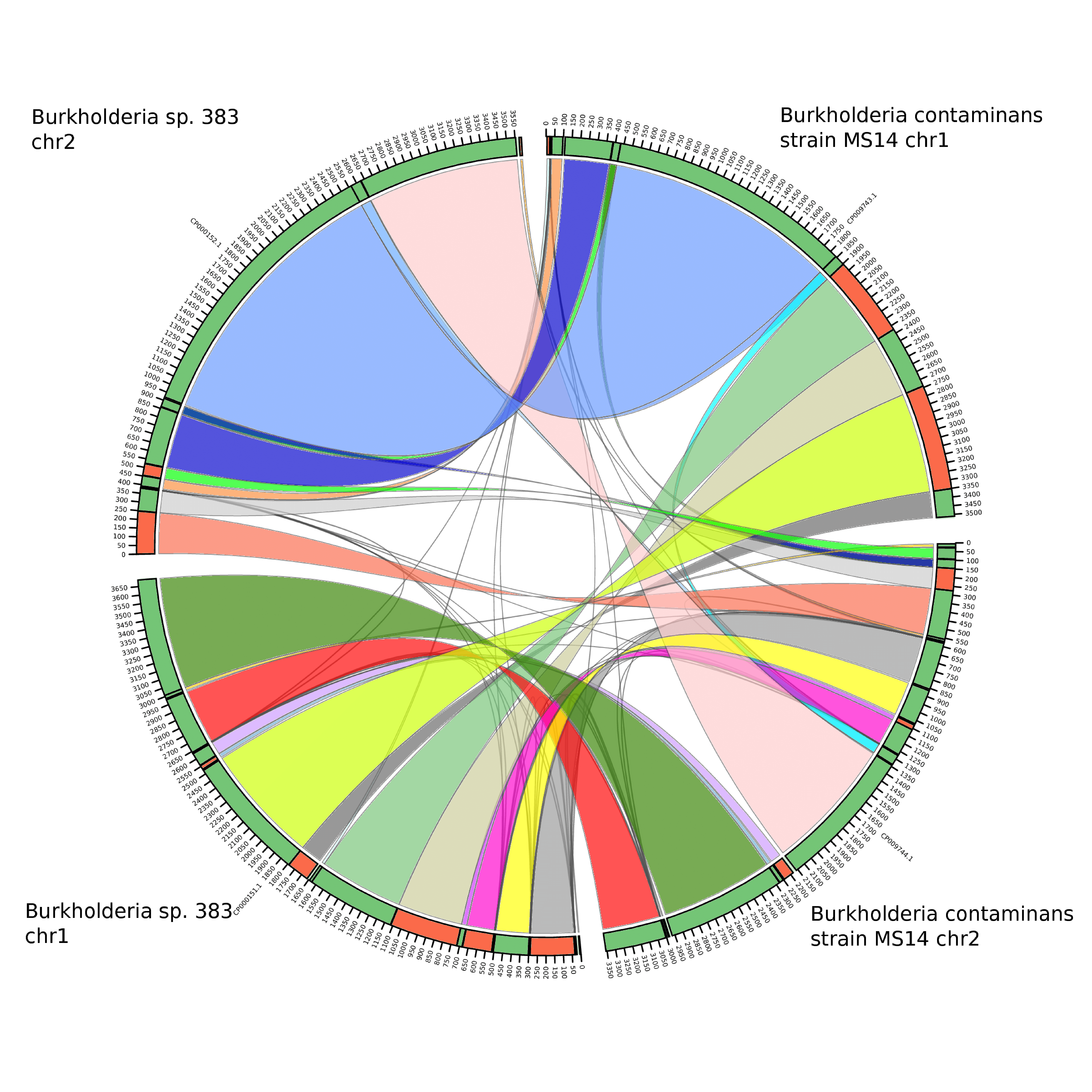
The project aims to understand organization and evolutionary benefits of bacterial genomes with secondary replicons. Most bacterial genomes have a single chromosome that may be supplemented by a few smaller, dispensable plasmids. But approximately 10% of the bacteria with completely sequenced genome, mostly pathogens and plant symbionts, have essential megaplasmids and/or chromids. However, the advantages of multichromosomal genome organization remain unclear.
The project was supported by the Marie SkłodowskaCurie Grant No. 754411.
Evolution of virulence factors in human-host and non-human-host invasive Escherichia
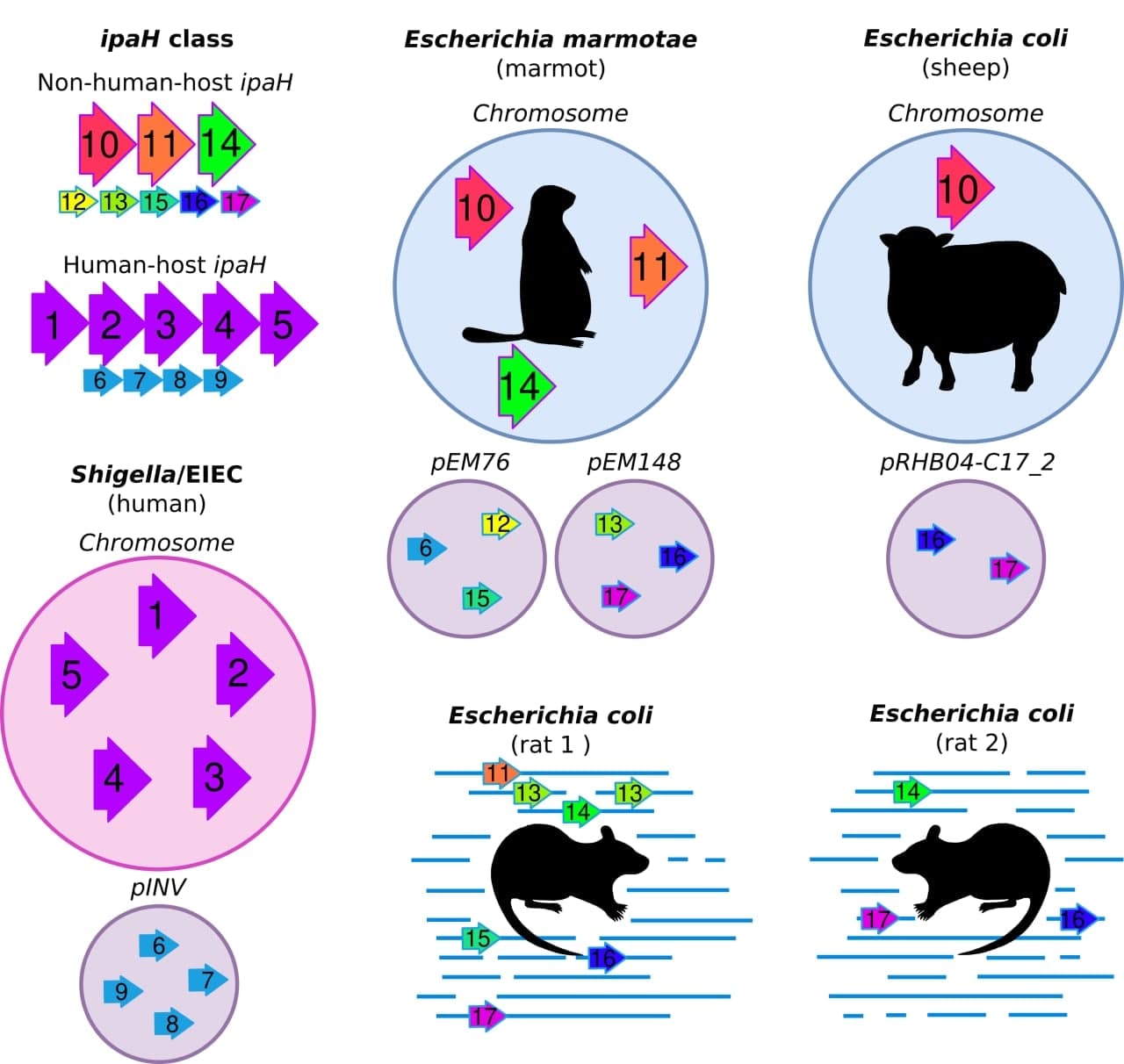
The project aims to describe composition and evolution of ipaH genes, effectors of Type 3 secretion system, in pathogens of different hosts. These genes are key factors of Shigella invasion that which are used for disease genotyping. Until recently, Shigella were thought to be primate-restricted pathogens. However, recent genomic studies confirmed ipaH genes in genome of Escherichia marmotae, a potential marmot pathogen, and of an E. coli extracted from fecal samples of bovine calves, suggesting that non-human hosts may also be infected by these potentially pathogenic to humans strains. We employ a computational approach to predict whether different Escherichia may also be an infectious agent of non-human hosts, which, therefore, may serve as a reservoir of human pathogens and virulence genes.
Сooperation partners:
2023-present Prof. David Berry, University of Vienna, Vienna, Austria. Project: Identification of shafflons in gut microbiomes. 2022-present Prof. Fyodor Kondrashov, OIST, Okinawa, Japan. Project: Selection forces in multipartite bacterial genomes. 2022-present Prof. Calin Guet IST Austria, Klosterneuburg, Austria. Project: Conservation of non-canonical start codons in mar operon genes. 2022-present Prof. Olga Kalinina, Helmholtz Institute for Pharmazeutical Research Saarland (HIPS), Germany. Project: Machine learning and phylogenetic analysis improves predicting antibiotic resistance in M. tuberculosis 2021-present Prof. Dan I. Andersson, Uppsala University, Sweden. Project: Fitness effects of short random peptides. 2021-present Prof. Mikhail Gelfand, IITP RAS, Russia. Project: Evolution of multi-partite genomes. 2020-2022 Dr. Nikita Alexeev, ITMO University, Saint Petersburg, Russia. Project: Development of the bioinformatic toolkit for whole-genome analysis. 2020-2022 Prof. Christoph Gasche, Medical University of Vienna, Vienna, Austria. Project: Genetic factors under biofilms formation in pathogenic E.coli. 2016-2018 Prof. Marc Robinson-Rechavi, Evolutionary Bioinformatics Lab, Department of Ecology and Evolution, Université de Lausanne, Lausanne, Switzerland. Project: Positive selection and horizontal gene transfer in prokaryotes. 2010-2014 Prof. Pavel Pevzner, University of California at San Diego, California, USA. Project: Application of the MGRA (Multiple Genome Rearrangements and Ancestors) algorithm to microbial data.